Galaktozemia a nietolerancja laktozy
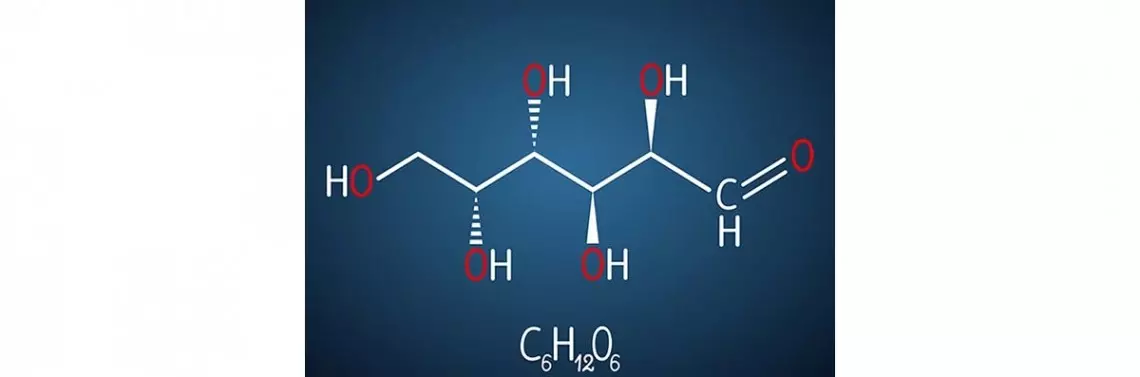
Na początku warto poświęcić kilka słów laktozie, aby wyjaśnić różnicę między galaktozemią a nietolerancją laktozy. To inaczej cukier mleczny, składający się z D-glukozy i D-galaktozy, które połączone są ze sobą wiązaniem beta-1,4-glikozydowym. W naturalnej postaci znajduje się tylko w mleku ssaków, z wyjątkiem lwów morskich, u których nie stwierdza się jej zawartości lub jest ona znikoma [3]. Laktoza, oprócz źródła energii, spełnia kilka korzystnych funkcji dla organizmu, m.in. ma właściwości prebiotyczne, wpływając tym samym na motorykę jelit. Ponadto jest mniej słodka od glukozy i sacharozy, co wpływa na regulację łaknienia, a także prawidłowy rozwój smaku u noworodków. Z kolei jej składowa – galaktoza – jest ważnym cukrem wchodzącym w skład ośrodkowego układu nerwowego. W celu spełnienia tych funkcji laktoza musi ulec hydrolizie w jelicie cienkim przez enzym laktaza, znajdujący się w najwyższym stężeniu w glikokaliksie rąbka szczoteczkowego kosmków jelitowych [4]. Znanych jest kilka typów nietolerancji laktozy, np. hipolaktazja typu dorosłych czy wtórna nietolerancja laktozy. W schorzeniu tym wyróżnia się również wrodzony brak laktazy (alaktazja). Jest to jednak bardzo rzadka przypadłość, gdyż dotychczas opisano zaledwie kilkadziesiąt przypadków tej choroby w Finlandii [5]. W tej sytuacji chorzy nie tolerują laktozy nawet...
Pozostałe 90% treści dostępne jest tylko dla Prenumeratorów

- Roczną prenumeratę dwumiesięcznika Food Forum w wersji papierowej lub cyfrowej,
- Nielimitowany dostęp do pełnego archiwum czasopisma,
- Możliwość udziału w cyklicznych Konsultacjach Dietetycznych Online,
- Specjalne dodatki do czasopisma: Food Forum CASEBOOK...
- ...i wiele więcej!
Dołącz do grona dietetyków i specjalistów świadomego żywienia
Co dwa miesiące otrzymuj gotowe rozwiązania, aktualne badania i sprawdzone terapie, które możesz od razu wykorzystać w praktyce. Rozwijaj swoje kompetencje, pracuj skuteczniej i bądź na bieżąco z najnowszymi trendami żywieniowymi i innowacjami w dziedzinie dietetyki.